Computational Study of Amino Mediated Molecular Interaction Evidenced in N 1s NEXAFS: 1,4-Diaminobenzene on Au (111)
Abstract
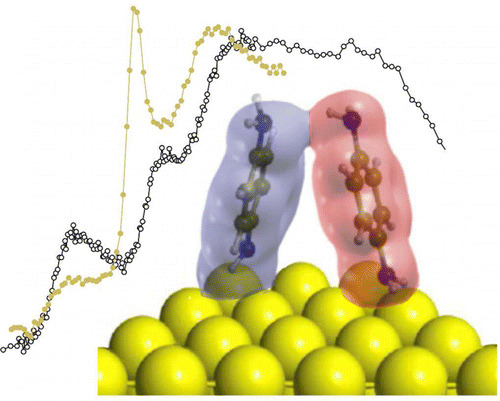
Primary amines can interact with neighbor molecules or with a metal substrate via weak bonds involving the electron lone pair of their amino functional group. Near edge X-ray absorption spectra (NEXAFS) on the N 1s edge show that the structure of the empty molecular orbitals localized on the nitrogen atom is very sensitive to these interactions. Here we investigate the origin of these changes by means of theoretical calculations. NEXAFS spectra are simulated for the 1 4-benzenediamine (BDA) molecule in its free, crystalline, and monolayer on Au(111) forms. We identify the electronic states which are affected by these amino-based interactions. In the case of the molecular layer grown on the gold substrate, we show how the results of the calculations can be used to identify intermolecular interactions influencing adsorption geometries in molecular monolayers.